




合作客戶/
拜耳公司 |
同濟大學(xué) |
聯(lián)合大學(xué) |
美國保潔 |
美國強生 |
瑞士羅氏 |






相關(guān)新聞Info
-
> 乳化瀝青穩(wěn)定性影響因素
> 4種油醇烷氧基化物平衡和動態(tài)表面張力、潤濕性、泡沫性、乳化性質(zhì)研究(三)
> 3種典型清水劑對不同原油組分界面穩(wěn)定性、油滴聚并行為的影響(一)
> 浮選藥劑的性能、組合用藥機理及協(xié)同效應(yīng)的影響因素(一)
> 新型助排劑配方組分、對表/界面性能的影響及助排效果(三)
> 礦井瓦斯防治:表面活性劑溶液表面張力、泡沫特性及對甲烷緩釋效應(yīng)(二)
> 應(yīng)用熒光顯微鏡研究了蛋白質(zhì)在氣-水界面的組裝——結(jié)果和討論
> Langmuir-Blodgett法制備環(huán)糊精單分子或多分子層膜
> 不同濃度下甘草酸溶液界面張力的變化
> 有效勘測地下水污染我們在行動!
推薦新聞Info
-
> 系列脂肪醇聚氧乙烯醚磺酸鹽表面活性劑制備、溶解性、表面張力及界面張力測定(二)
> 系列脂肪醇聚氧乙烯醚磺酸鹽表面活性劑制備、溶解性、表面張力及界面張力測定(一)
> 添加不同量阿維菌素Silwet 408對阿維菌素微乳劑藥液表面張力的影響——結(jié)果與分析、結(jié)論
> 添加不同量阿維菌素Silwet 408對阿維菌素微乳劑藥液表面張力的影響——摘要、材料與方法
> 新型均相微乳液型助排劑AO-4表/界面張力測定及室內(nèi)評價——結(jié)果與討論、結(jié)論
> 新型均相微乳液型助排劑AO-4表/界面張力測定及室內(nèi)評價——摘要、實驗部分
> 正己醇聚氧乙烯醚硫酸鈉、正己醇聚氧丙烯醚硫酸鈉水溶液平衡表面張力、動態(tài)表面張力測定(二)
> 正己醇聚氧乙烯醚硫酸鈉、正己醇聚氧丙烯醚硫酸鈉水溶液平衡表面張力、動態(tài)表面張力測定(一)
> 遼河油田原油的石油酸、石油堿組分萃取過程、結(jié)構(gòu)表征及界面張力測量——結(jié)果與討論、結(jié)論
> 遼河油田原油的石油酸、石油堿組分萃取過程、結(jié)構(gòu)表征及界面張力測量——實驗部分
St與MMA在無皂乳液聚合過程中的動態(tài)表面張力變化——結(jié)果與討論、結(jié)論
來源: 《化工學(xué)報》 瀏覽 790 次 發(fā)布時間:2024-11-01
2、結(jié)果與討論
2.1無皂乳液聚合體系組分的動態(tài)表面張力未反應(yīng)前,聚合體系可以分為油相和水相兩部分:油相部分主要為單體,水相部分主要為引發(fā)劑(過硫酸鉀)、pH緩沖劑(NaHCO。)、PEG和水。這兩部分的DST曲線如圖1所示。
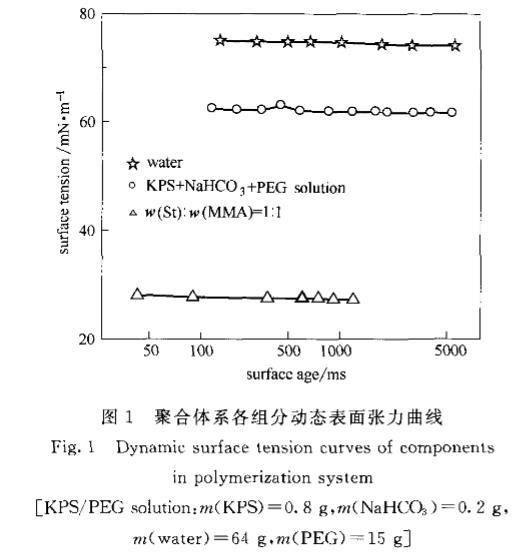
圖1聚合體系各組分動態(tài)表面張力曲線
從圖中可以看出,油相中混合單體的氣液界面張力最低(約25mN·m左右),水的表面張力最高(約74mN·m左右),而受引發(fā)劑、緩沖劑和PEG的影響,水相混合溶液的氣液界面張力居中(約62mN·m左右)。在未反應(yīng)之前,水油兩相的DST曲線變化趨勢很小,都能以較快的速度達到平衡穩(wěn)定狀態(tài)。
2.2聚合時間對動態(tài)表面張力變化趨勢的影響經(jīng)引發(fā),水相中溶解的單體聚合成鏈,聚合體系的組分也隨之發(fā)生變化。體系在水相中含有了自由基齊聚物,以及由齊聚物聚集形成的基本粒子或膠束。這些新產(chǎn)生的物質(zhì)對DST曲線有很明顯的影響,加上PEG與聚合物鏈的相互作用,則使DST曲線有更多的變化。為了能明確地分析PEG在聚合過程中的作用,進行了有PEG和無PEG參與的聚合體系各時間段動態(tài)表面張力實驗比較。圖2和圖3分別為St/MMA在無PEG的水中和有PEG的水溶液中DST隨聚合時間的變化趨勢。
由圖2可見,無PEG參與的聚合體系中,隨聚合進行,體系所表現(xiàn)的氣液界面張力趨勢為逐漸增大,從5min的28mN·m~左右到6Omin的60~65ITIN·m~,75min則稍有回落,在55~61mN·m。各時間段的DST趨勢較為平穩(wěn)。這種氣液界面張力隨聚合進行逐漸上升的趨勢較為符合無皂乳液聚合反應(yīng)機理。在反應(yīng)初期水相中不僅有溶解在水中的單體,還有引發(fā)形成的齊聚物,這種齊聚物一端帶有疏水鏈段,另一端帶有親水性的一SO引發(fā)劑碎片,具有表面活性。當(dāng)齊聚物達到膠束濃度時會聚并成基本粒子,由幾十個到上百個齊聚物組成。溶解在水相中的單體分子量相對齊聚物小很多,體積小,擴散速率快,極易遷移至氣液界面。對于一開始的反應(yīng),體系中仍有大量單體存在,水相中的單體濃度基本保持在飽和狀態(tài),因此大部分氣液界面首先由單體等物質(zhì)占據(jù),還有極少部分的界面由具有表面活性的齊聚物覆蓋,如圖4所示。所以與圖1對比會發(fā)現(xiàn),5min時的氣液界面張力非常接近單體本身的氣液界面張力。隨聚合進行,水相中的單體逐漸耗光,氣液界面張力開始逐漸上升,此時到達氣液界面的則主要是齊聚物和膠粒,相對單體的遷移速率要慢很多,因此10min以后的DST衰減斜率要大于5rain時的。
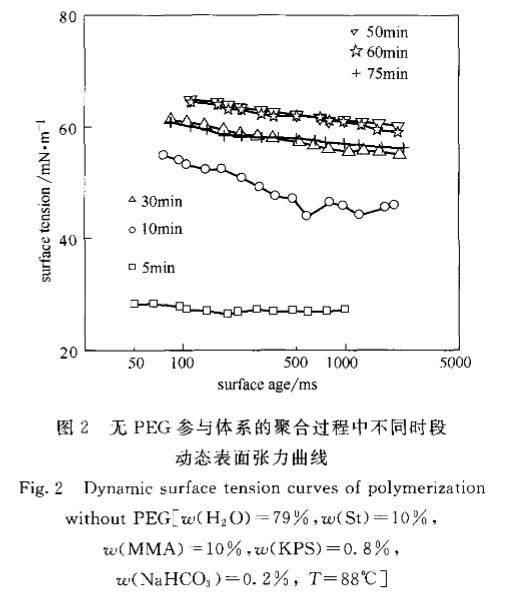
圖2無PEG參與體系的聚合過程中不同時段動態(tài)表面張力曲線
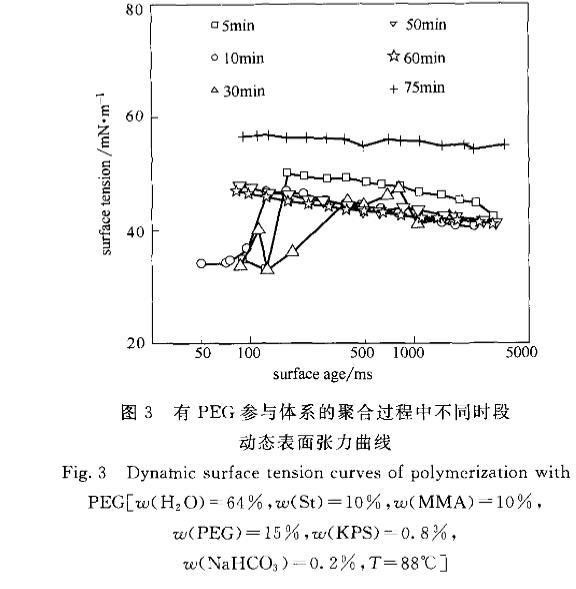
圖3有PEG參與體系的聚合過程中不同時段動態(tài)表面張力曲線
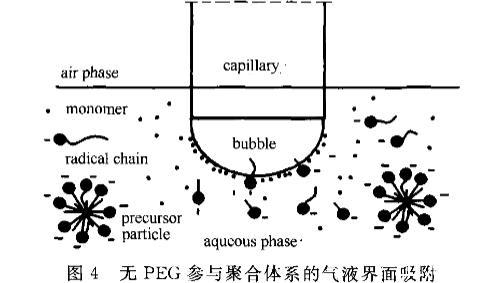
圖4無PEG參與聚合體系的氣液界面吸附
由圖3可見,有PEG參與的聚合體系。5rain的DST與圖2相比變化起伏較大,有先低后急升再緩降的變化模式,與一般的DST呈單調(diào)減小的趨勢有很大不同。這是因為在反應(yīng)剛開始體系中除了單體、齊聚物和膠粒,又增加了PEG,而且PEG與齊聚物、膠粒作用形成粒子堆口]。PEG是完全水溶性物質(zhì),在體系水相中的濃度遠遠大于單體和齊聚物,但擴散速率要小于單體及齊聚物,因此一開始部分新形成的氣液界面會由擴散速率最快的單體迅速占領(lǐng),而隨時間推移PEG借助濃度優(yōu)勢會替代單體占據(jù)氣液界面,如圖5所示,最終達到平衡,由單體、齊聚物和PEG共同覆蓋整個氣液界面。PEG水溶液的氣液界面張力要比單體的氣液界面張力大得多,因此在水相中有大量單體存在的情況下,此聚合體系的DST會出現(xiàn)先低后急升的曲線形狀。這個狀況會一直持續(xù)到30rain時,可以看出,PEG使體系直到30min時仍有大量游離單體存在于水相,而無PEG參與的體系此時已無單體游離在水相中。這一點可以從圖6轉(zhuǎn)化率一時間曲線得到證實,無PEG參與的體系30min時已接近聚合終點,而有PEG參與的體系轉(zhuǎn)化率才只有25左右。待反應(yīng)至50min時,DST趨勢平穩(wěn),水相中游離的單體已經(jīng)很少,此時轉(zhuǎn)化率在49左右,說明單體已基本進入到膠粒中。這個狀態(tài)一直維持到60min時。75min時的體系有很大變化,從宏觀看粒徑發(fā)生轉(zhuǎn)變,由大變小。從DST曲線看氣液界面張力明顯提高,轉(zhuǎn)化率已快速增加到95,接近終點。此時體系中對DST的貢獻主要以脫離粒子堆的成熟粒子l3和PEG為主。

圖5有PEG參與聚合體系的氣液界面吸附
2.3 St/M MA無皂乳液聚合物的表面活性
圖2和圖3的DST針對混合體系,只能說明PEG如何影響聚合反應(yīng)。但對最終聚合物的特性是否發(fā)生變化還無法判斷。為了進一步說明PEG如何影響聚合物特性,將PEG、有PEG參與聚合的10min和75min共聚物以及無PEG參與聚合體系的共聚物按1.4節(jié)中的制備聚合物乳液樣品的方法制備聚合物乳液,然后測試DST,得到圖7。
由圖7可見,有PEG參與的聚合體系制備的聚合物乳液表面張力較低(約5O~51mN·m左右),明顯低于四氫呋喃/水、PEG/四氫呋喃/水、無PEG參與的聚合體系制備的聚合物乳液表面張力(約54~55.5mN·rfl左右)。因為氣液界面張力受擴散物質(zhì)本身表面活性強弱的影響,所以從四氫體系制備的聚合物乳液DST相近可以得出結(jié)論,即T≤PE。、F≤EG聚合物,而有PEG參與的聚合物乳液的界面張力說明c聚合物<n,說明有PEG參與的聚合體系制備的聚合物表面活性更強,而且這種較強表面活性的聚合物在反應(yīng)的開始就已經(jīng)形成。
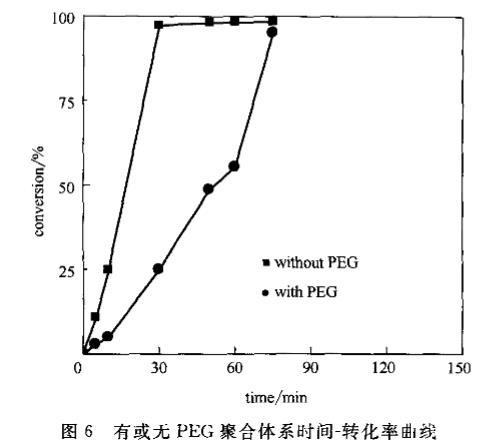
圖6有或無PEG聚合體系時間一轉(zhuǎn)化率曲線
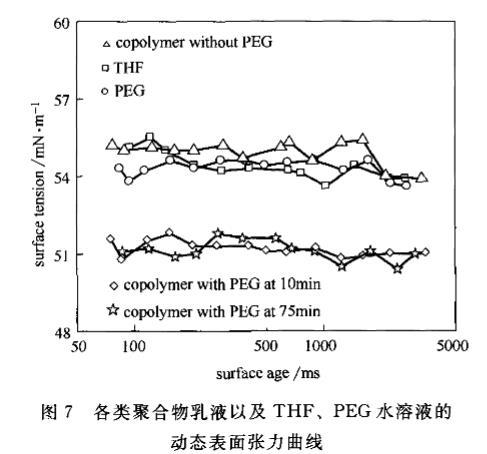
圖7各類聚合物乳液以及THF、PEG水溶液的動態(tài)表面張力曲線
由于開始轉(zhuǎn)化率較低,強表面活性的聚合物生成量較少,形成的聚合物團聚成較大的粒子堆,這時粒徑表現(xiàn)是由小變大。隨轉(zhuǎn)化率的不斷增加,強表面活性聚合物的量大幅增加,這種較強表面活性的聚合物更易形成粒徑小的穩(wěn)定膠粒,不斷進人粒子堆中的粒子形成粒徑小且密實的粒子,使粒子的表面電荷增加,并最終脫離粒子堆形成穩(wěn)定粒子,這時粒徑表現(xiàn)是由大變小。PEG參與的體系聚合物所具有的強表面活性正是聚合物不斷進入粒子的動力,證實了所觀察到的體系粒徑由小變大、再由大變小的現(xiàn)象。
3、結(jié)論
由于PEG的存在,使St/MMA在水相中經(jīng)歷了不一樣的聚合過程,PEG會使體系的反應(yīng)速率變慢,當(dāng)反應(yīng)到50min時轉(zhuǎn)化率只有49,而無PEG參與的體系在30min時聚合已接近終點,轉(zhuǎn)化率達到98以上。St/MMA和引發(fā)劑形成的聚合物在PEG的作用下,形成了一種具有更強表面活性的聚合物,其表面活性水平高于PEG以及無PEG參與的聚合體系的聚合物,但弱于混合單體及齊聚物。這種較強表面活性的聚合物在轉(zhuǎn)化率只有5的時候就已經(jīng)存在,是聚合物進入粒子堆形成小粒子且穩(wěn)定粒子的動力,并最終使粒子帶有足夠脫離粒子堆的表面電荷。